Tree Arithmetic with Phylogenetic Flows
phyf_arith.Rmd
library(phyf)
#>
#> Attaching package: 'phyf'
#> The following object is masked from 'package:stats':
#>
#> pf
library(ape)
library(withr)In this vignette we will see how the phyf package
makes it easy to manipulate phylogenetic tree by performing arithmetic
on phylogenetic flow collection objects (pfc). A
phylogenetic flow collection is a way to represent a rooted phylogenetic
as a set of ‘flows’ from the root of the tree to its terminal nodes
(e.g. the tree’s tips). Each edge between the root and the tips can have
data associated with it, which we call phylogenetic flow features
(pff). Often these feature will be the branch length
associated with the edge. We can perform arithmetic on these features to
do lots of interesting and useful thing. In this vignette, we will focus
on using pfc arithmetic to perform the classic branch
length transformations known as Pagel’s transformations. There are
three: ‘lambda’, ‘delta’, and ‘kappa’.
Pagel’s Kappa
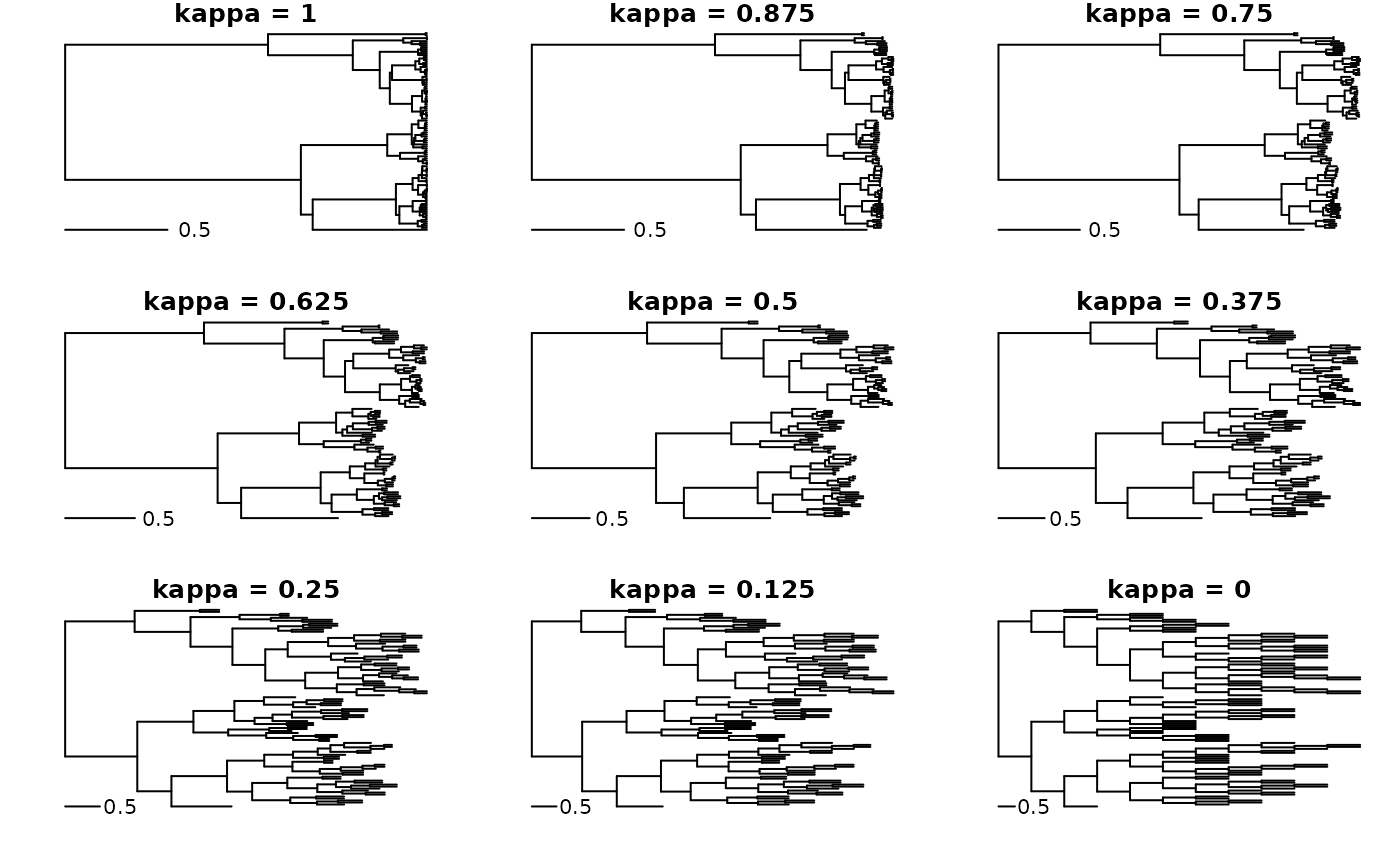
We will start with the kappa transformation because it is the
simplest one from the perspective of a phylogenetic flow. It is simply
raising the branch lengths of a phylogenetic tree to some power (which
is the kappa parameter). Typically, kappa values less than one are used,
but values greater than one are also possible. Kappa values less than
one tend to ‘shrink’ branch lengths non-linearly towards 1, until we
have at kappa = 0, all branch lengths are exactly one. This is sometimes
used to model ‘speciational’ change along branches, because any model
weighted by the branch lengths will be insensitive to branch length,
only the branching structure itself. First we start by generating a
random tree using ape, converting that to a
pfc. Then we demonstrate the transformation.
When we create a pfc object from a phylo
object, the branch lengths of the tree are automatically used as the
phylogenetic flow feature. We can easily perform a kappa transformation
by directly raising the pfc object to the desired kappa
parameter. Let’s test it with kappa = 0.5
tree_pfc_0.5 <- tree_pfc^0.5
plot(tree_pfc_0.5)
We can see as a result of the transformation that longer branches have become relatively shorter compared with branches that started out shorter. We can do a series of transformation to see how the tree converges to kappa = 0.
with_par(list(mfrow = c(3, 3), mar = c(2, 2, 1, 1), xpd = NA), {
for(i in seq(1, 0, length.out = 9)) plot(tree_pfc^i, main = sprintf("kappa = %s", i), show.tip.label = FALSE)
})
Pagel’s lambda and delta transformations are a little more complicated but still pretty easy to calculate in phyf.
Pagel’s Lambda
Pagel’s lambda transformation multiplies the internal branch lengths by a constant (the lambda parameter), whilst compensatorily growing the terminal branches to keep the total distances from the root to the tips the same. Again, lambda is usually set to less than 1, such that as lambda goes to zero the internal branches shrink to nothing, leaving only the terminla branches. This model is derived by transforming the phylogenetic variance-covariance matrix implied by a phylogeny. Lambda > 1 is not usually used because the resulting variance-covariance matrix can become ill-conditioned. From the perspective of branch length transformation this is because if you multiply an internal branch by two much greater than zero, its height can become greater than the height of the tips, and so you cannot maintain the total distance from the root to tips without negative branch lengths!
So to do the transformation in phyf we need to do two transformations, one on the internal branches and one on the external branches. We also need information on the root to tips lengths, which is the sum of each phylogenetic flow feature.
lambda <- 0.5
lambda_pfc <- tree_pfc
root2node_lens <- pf_flow_sum(lambda_pfc)
pf_internal(lambda_pfc) <- pf_internal(lambda_pfc) * lambda
pf_terminal(lambda_pfc) <- pf_terminal(lambda_pfc) + (root2node_lens - pf_flow_sum(lambda_pfc))
plot(lambda_pfc, show.tip.label = FALSE)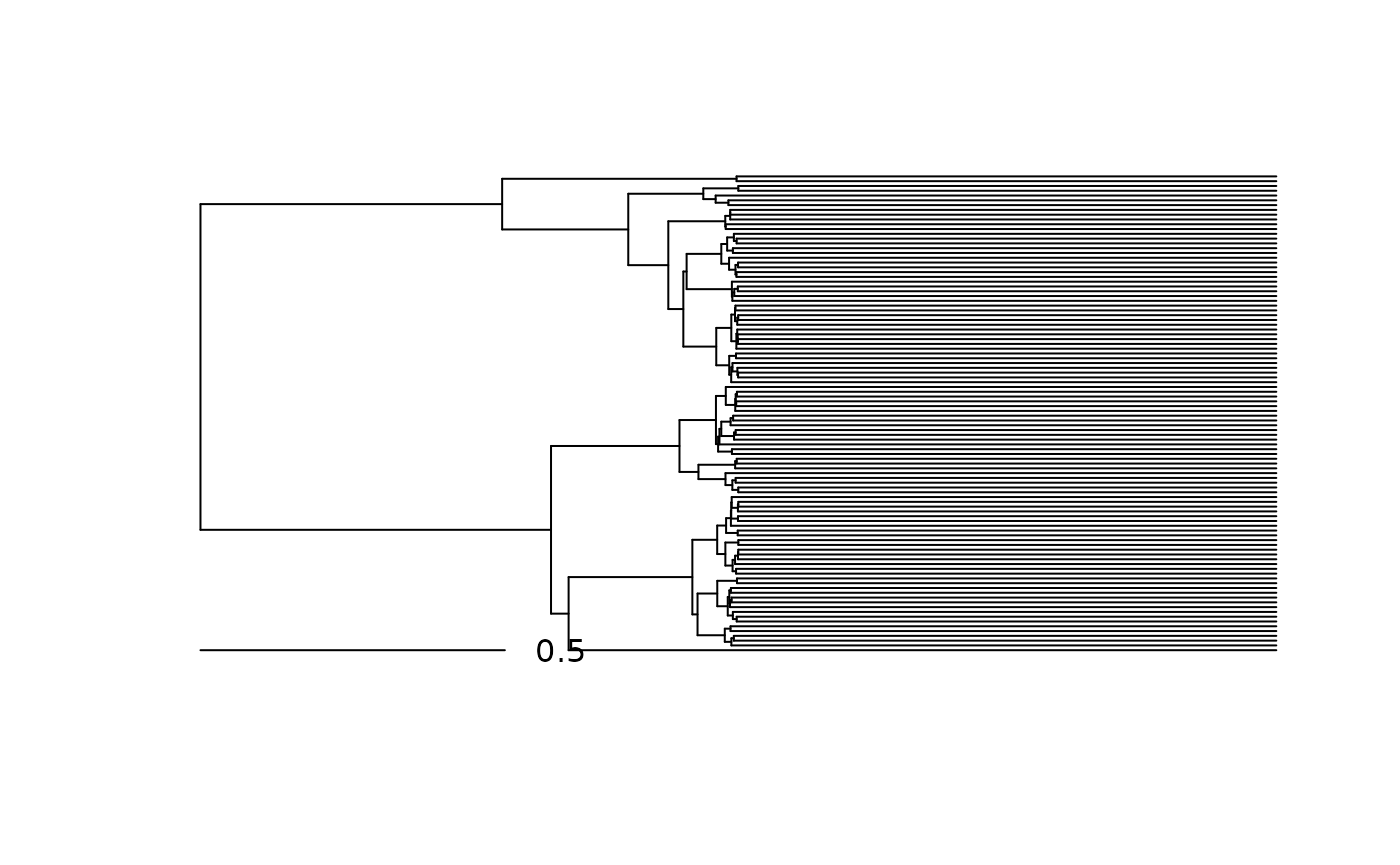
lambda_trans <- function(x, lambda = 0.5) {
root2node_lens <- pf_flow_sum(x)
pf_internal(x) <- pf_internal(x) * lambda
pf_terminal(x) <- pf_terminal(x) + (root2node_lens - pf_flow_sum(x))
x
}
with_par(list(mfrow = c(3, 3), mar = c(2, 2, 1, 1), xpd = NA), {
for(i in seq(1, 0, length.out = 9)) plot(lambda_trans(tree_pfc, i), type = "f", main = sprintf("lambda = %s", i), show.tip.label = FALSE)
})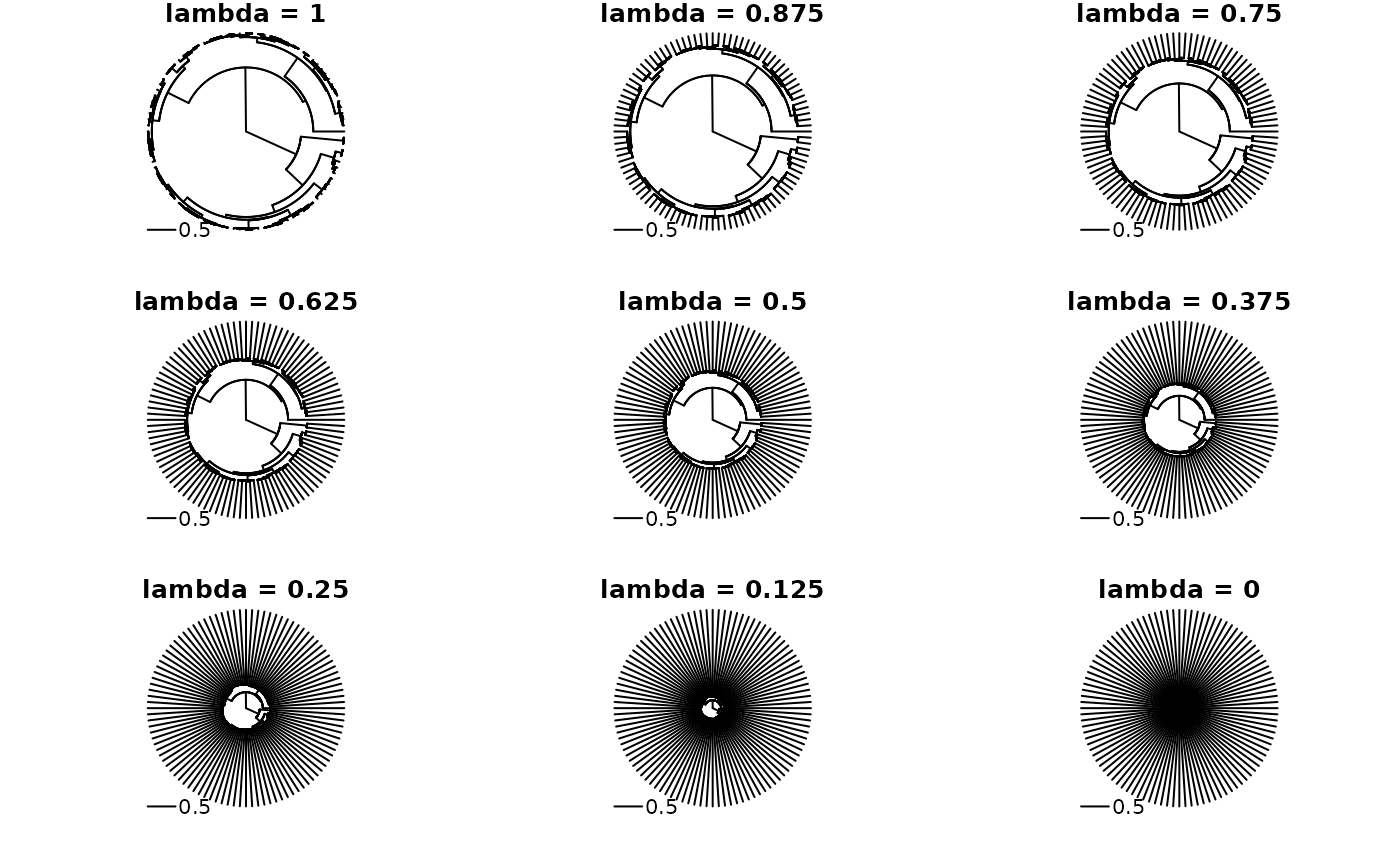 ## Pagel’s Delta
## Pagel’s Delta
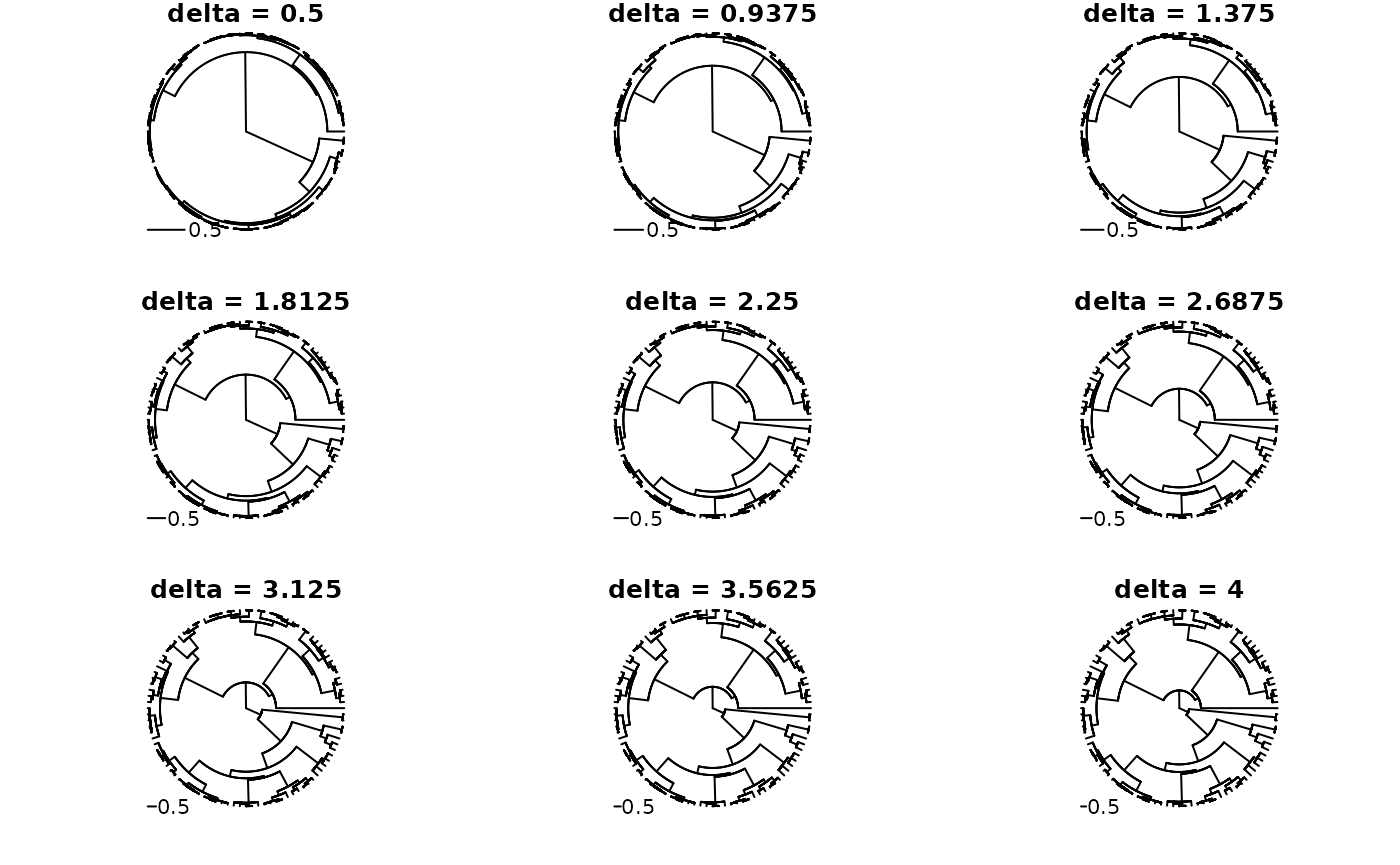
Lastly, Pagel’s delta transformation is a transformation of the node
heights of a phylogeny, raising each to the power of the delta
parameter. This has the effect of shrinking branches near the tips more
than those near the root. In the context of a model of evolution on the
tree, this is used to model a ‘slow-down’ of evolution through time. We
can acheive this transformation by first generating a pfc
where the node heights are the phylogenetic flow features instead of the
branch lengths. Node heights can be calculated as the cumulative sum of
branch lengths down each phylogenetic flow. phyf has the
pf_flow_cumsum() function for this.
node_heights <- pf_flow_cumsum(tree_pfc)
head(node_heights)
#> <pfc<e:198>[6]>
#> First 6 phylogenetic flows:
#> [1] ◎── 1.2──→ Node3 ── 1.2──→ Node4 ── 1.8──→ t24
#>
#> [2] ◎── 1.2──→ Node3 ── 1.2──→ Node4 ── 1.6──→ Node10 ── 1.6──→ Node11 ── 1.7──→ Node23 ── 1.7──→ Node46 ── 1.8──→ t13
#>
#> [3] ◎── 1.2──→ Node3 ── 1.2──→ Node4 ── 1.6──→ Node10 ── 1.6──→ Node11 ── 1.7──→ Node23 ── 1.7──→ Node46 ── 1.8──→ Node58 ── 1.8──→ t7
#>
#> [4] ◎── 1.2──→ Node3 ── 1.2──→ Node4 ── 1.6──→ Node10 ── 1.6──→ Node11 ── 1.7──→ Node23 ── 1.7──→ Node46 ── 1.8──→ Node58 ── 1.8──→ t40
#>
#> [5] ◎── 1.2──→ Node3 ── 1.2──→ Node4 ── 1.6──→ Node10 ── 1.6──→ Node11 ── 1.7──→ Node23 ── 1.7──→ Node39 ── 1.8──→ t48
#>
#> [6] ◎── 1.2──→ Node3 ── 1.2──→ Node4 ── 1.6──→ Node10 ── 1.6──→ Node11 ── 1.7──→ Node23 ── 1.7──→ Node39 ── 1.8──→ t81
plot(node_heights)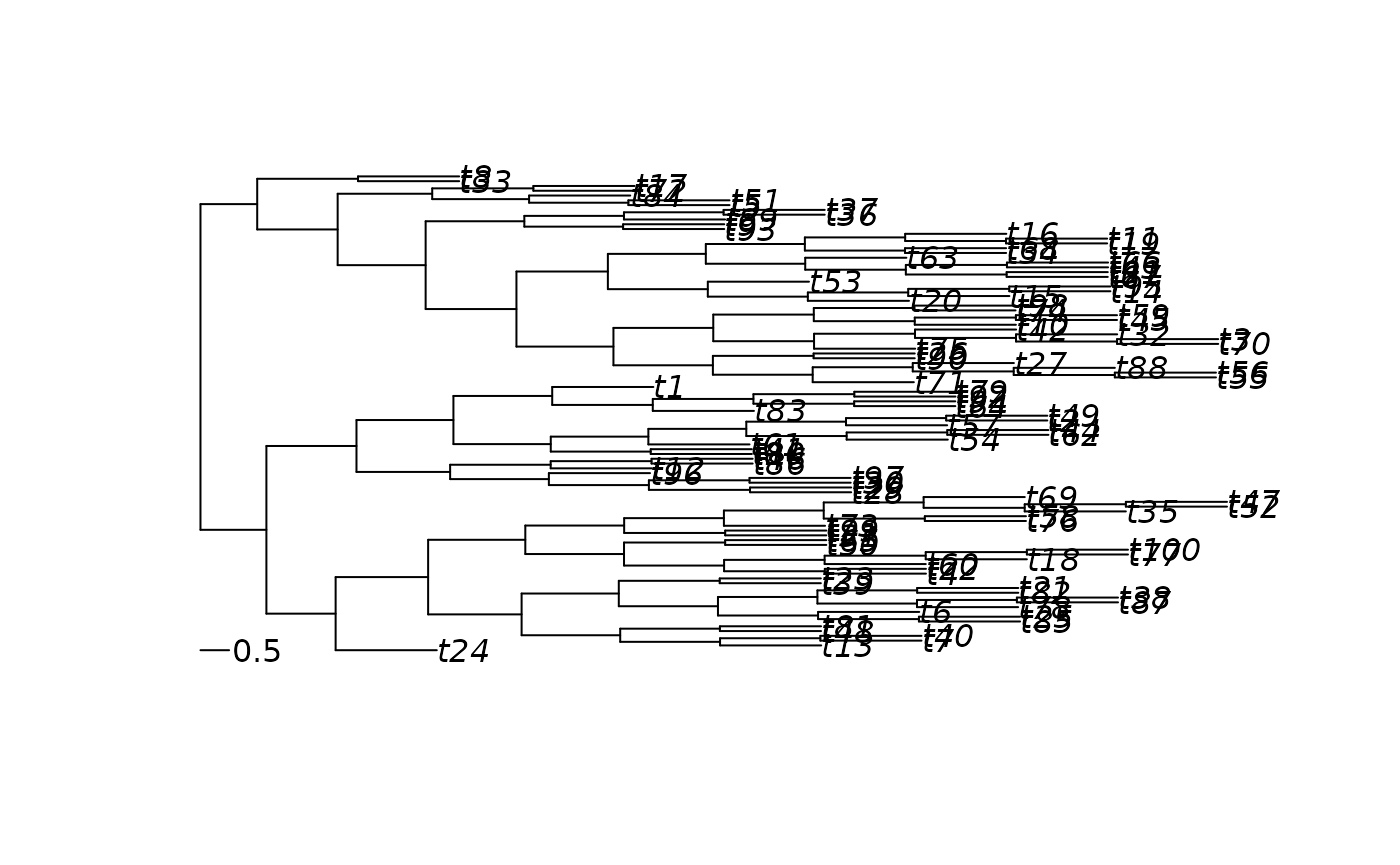
delta <- 0.4
new_pfc <- node_heights^deltaNow to get back branch lengths instead of node heights all we need to
do is subtract from each node height the node height of its immediate
ancestor. phyf also has a convenient function for this:
pf_anc(), which returns a pfc with each
feature replaces by the feature value of its ancestor.
head(pf_anc(new_pfc))
#> <pfc<e:198>[6]>
#> First 6 phylogenetic flows:
#> [1] ◎── 0.0──→ Node3 ── 1.1──→ Node4 ── 1.1──→ t24
#>
#> [2] ◎── 0.0──→ Node3 ── 1.1──→ Node4 ── 1.1──→ Node10 ── 1.2──→ Node11 ── 1.2──→ Node23 ── 1.2──→ Node46 ── 1.2──→ t13
#>
#> [3] ◎── 0.0──→ Node3 ── 1.1──→ Node4 ── 1.1──→ Node10 ── 1.2──→ Node11 ── 1.2──→ Node23 ── 1.2──→ Node46 ── 1.2──→ Node58 ── 1.3──→ t7
#>
#> [4] ◎── 0.0──→ Node3 ── 1.1──→ Node4 ── 1.1──→ Node10 ── 1.2──→ Node11 ── 1.2──→ Node23 ── 1.2──→ Node46 ── 1.2──→ Node58 ── 1.3──→ t40
#>
#> [5] ◎── 0.0──→ Node3 ── 1.1──→ Node4 ── 1.1──→ Node10 ── 1.2──→ Node11 ── 1.2──→ Node23 ── 1.2──→ Node39 ── 1.2──→ t48
#>
#> [6] ◎── 0.0──→ Node3 ── 1.1──→ Node4 ── 1.1──→ Node10 ── 1.2──→ Node11 ── 1.2──→ Node23 ── 1.2──→ Node39 ── 1.2──→ t81
new_pfc <- new_pfc - pf_anc(new_pfc)
plot(new_pfc)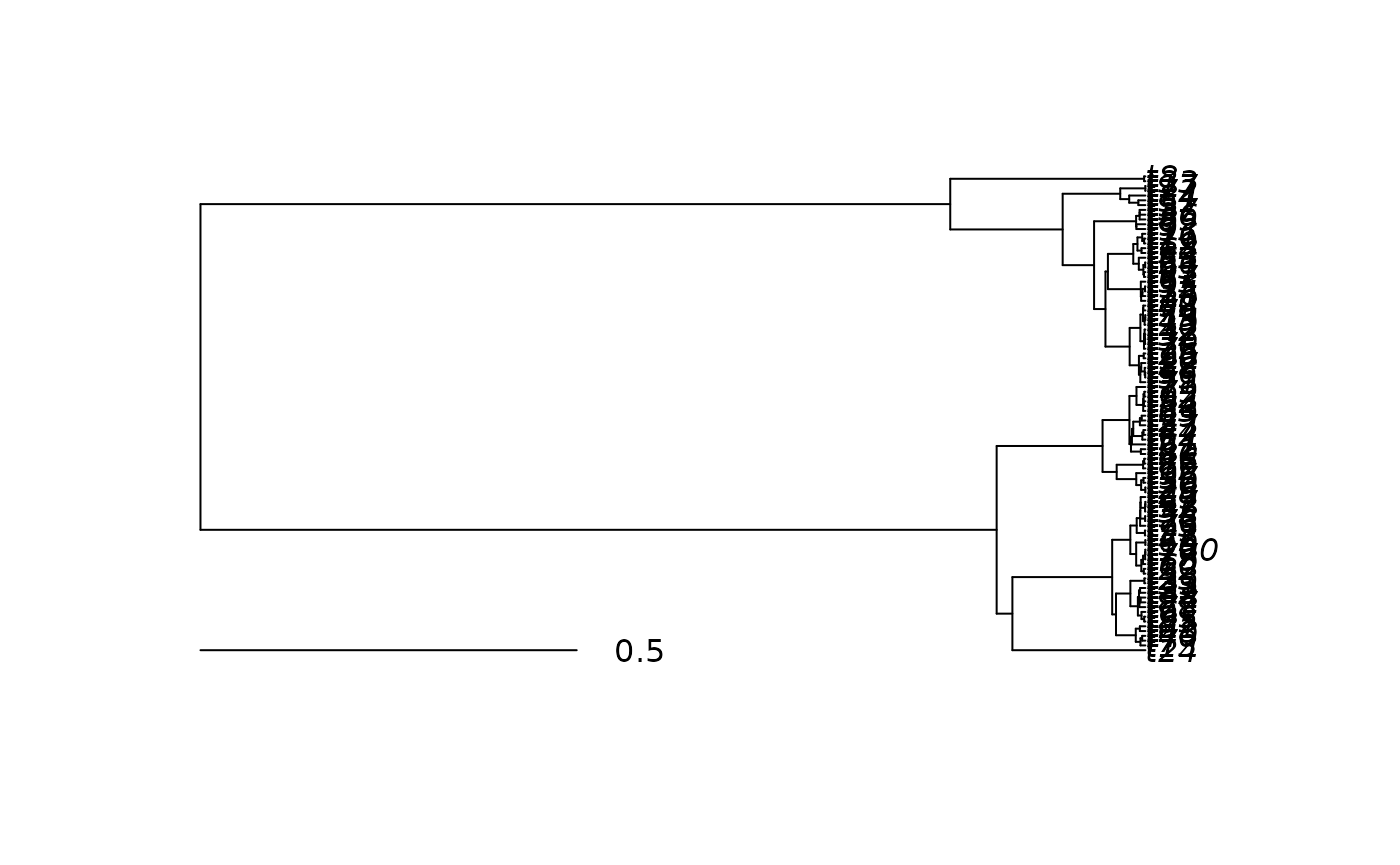
A series of deltas:
delta_trans <- function(x, delta = 0.5) {
new_pfc <- pf_flow_cumsum(tree_pfc)^delta
new_pfc <- new_pfc - pf_anc(new_pfc)
new_pfc
}
with_par(list(mfrow = c(3, 3), mar = c(2, 2, 1, 1), xpd = NA), {
for(i in seq(0.5, 4, length.out = 9)) plot(delta_trans(tree_pfc, i), type = "f", main = sprintf("delta = %s", i), show.tip.label = FALSE)
})