Introduction to slimr: Writing SLiM scripts from R
Source:vignettes/slimrlang_intro.Rmd
slimrlang_intro.RmdThis vignette introduces the slimr package.
slimr has the goal of making it easy to write scripts for
the population genetics simulation software and programming language SLiM. slimr offers
tools to get R objects and generated model parameters into SLiM, as well
as to help SLiM generate R friendly output for post-simulation analysis.
It also provides tools for working with existing SLiM scripts as well as
running, monitoring, and visualising SLiM simulations in realtime and
directly from R, as well as post-processing in R.
Writing SLiM Scripts
To write a SLiM script in slimr you must wrap your SLiM
code in slim_block function calls, which are then nested in
a slim_script function call. The API for slimr
has been designed to mimic as close as possible the way that scripts are
written in SLiM itself. To get you started we will use an example. SLiM
has an extremely extensive manual that includes a large number (around
150!) of example scripts to help users learn the language. We will take
advantage of this resource and try to write a few of the manual’s
scripts using the slimr approach.
This is the first SLiM script (referred to as ‘recipes’) in the SLiM manual:
// set up a simple neutral simulation
initialize()
{
// set the overall mutation rate
initializeMutationRate(1e-7);
// m1 mutation type: neutral
initializeMutationType("m1", 0.5, "f", 0.0);
// g1 genomic element type: uses m1 for all mutations
initializeGenomicElementType("g1", m1, 1.0);
// uniform chromosome of length 100 kb
initializeGenomicElement(g1, 0, 99999);
// uniform recombination along the chromosome
initializeRecombinationRate(1e-8);
}
// create a population of 500 individuals
1
{
sim.addSubpop("p1", 500);
}
// run to generation 10000
10000
{
sim.simulationFinished();
}So how would we write this script in slimr? Like
this:
library(slimr)
#> Welcome to the slimr package for forward population genetics simulation in SLiM. For more information on SLiM please visit https://messerlab.org/slim/ .
#>
#> Attaching package: 'slimr'
#> The following object is masked from 'package:methods':
#>
#> initialize
#> The following object is masked from 'package:base':
#>
#> interaction
slim_script(
slim_block(initialize(),
{
## set the overall mutation rate
initializeMutationRate(1e-7);
## m1 mutation type: neutral
initializeMutationType("m1", 0.5, "f", 0.0);
## g1 genomic element type: uses m1 for all mutations
initializeGenomicElementType("g1", m1, 1.0);
## uniform chromosome of length 100 kb
initializeGenomicElement(g1, 0, 99999);
## uniform recombination along the chromosome
initializeRecombinationRate(1e-8);
}),
slim_block(1,
{
sim.addSubpop("p1", 500);
}),
slim_block(10000,
{
sim.simulationFinished();
})
) -> script_1
script_1#> <slimr_script[3]>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 99999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }It is as simple as that at its most basic. slim_script
returns a slimr_script object which has a print method that
prints out the final SLiM script in a pretty fashion. You can see that
the output above is quite similar to the original SLiM code from the
manual recipe. The one difference is the block names, highlighted in
cyan. These are added internally by slimr to keep track of
code blocks. These names can be used in some other slimr
functions, for example, to refer to particular code blocks for
modification. You can also add block ‘ids’ inside of SLiM code, which
can be referred to within SLiM code. These are also fully supported by
slimr and are kept track of alongside the
slimr code block names. More on this later.
The SLiM language is similar enough to R that most of it works out of
the box without any modification. However, there are a few places that
SLiM is different enough that slimr has had to put in some
slight modifications. We will learn about these differences throughout
this vignette. At the moment however, let’s go through the typical
structure of a SLiM script and the slimr version of it.
Now, the script above is just the simplest way to write a SLiM script
in R, which is mostly just verbatim using the SLiM language syntax.
However, if you write your script this way, you will have to be very
familiar with the SLiM language, something which I personally am not
yet. In order to make it easier to write SLiM scripts in R,
slimr offers real-time code completion and SLiM function
documentation. In order to make this work within R however, you will
need to code your SLiM scripts slightly differently. This is a
consequence of the object-oriented approach of SLiM, which makes heavy
use of classes, methods and properties. Because R does not know the
classes of object which will only be determined after the script is run
within SLiM, you, the user, must specify what class an object is in
order for R to know what functions and properties are available to that
class, to enable autocompletion. To do this you can make use of the
special operator %.%, which mimics the .
operator within SLiM. SLiM, like Python, uses the .
operator to specify that a method or property after the .
belongs to the object before the .. It plays a similar role
to that of $ in R. An example of this in the above script
is these lines:
sim.addSubpop("p1", 500);and
sim.simulationFinished()In SLiM, sim is a global object of class
SLiMSim, which has a number of methods and properties
associated with it. You can rewrite the above statements in
slimr in the following way, which will then allow you to
use autocompletion:
sim%.%SLiMSim$addSubpop("p1", 500);and
sim%.%SLiMSim$simulationFinished();Simply replace the . with %.%, then specify
the class of object, in this case SLiMSim, followed by the
$ operator for R. The SLiMSim object contains
all the available methods and properties for its class. There is a
similar object for all SLiM classes. Note also that all SLiM class
objects in slimr have an abbreviated alias to save typing.
For SLiMSim this is SS, so you could type
instead sim%.%SS$simulationFinished(); for example. Using
this way of coding let’s you use autocomplete. Try typing just
sim%.%SS for example and (if using RStudio) you should see
this:
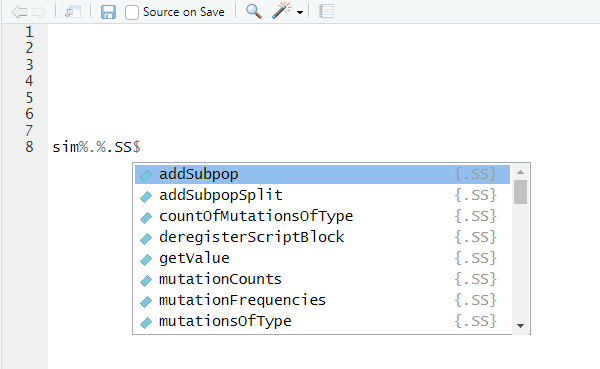
The popup gives you a list of all methods (and properties further
down) of the class. If you begin typing this list will be narrowed down
further. Once you’ve chosen a method to use, you can press the
tab key to bring up the arguments that the method accepts,
like this:
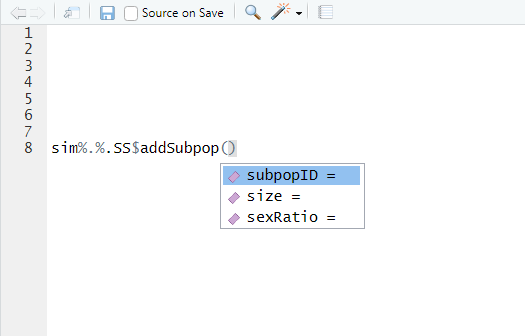
To get help on this method, including full descriptions of the
arguments, in RStudio, place you cursor over the function text and press
the F1 key. You can also simply run the function with no
arguments or type ?SS$addSubpop or
help("SS$addSubPop"), all of which will bring up the
documentation for this SLiM function.
help("SS$addSubpop")
#> No documentation for 'SS$addSubpop' in specified packages and librariesThere is also two special classes which are not actually classes in
SLiM itself, but they are used in slimr to keep particular
types of SLiM function together. These are Initialize (or
Init) and SLiMBuiltin (or SB).
The Initialize object contains SLiM functions that can only
be used in the initialize() block of SLiM code (more on
this later). SLiMBuiltin contains miscellaneous functions
built in to SLiM which are not part of any SLiM class.
Admittedly, using these extra R objects in the code does make it look
somewhat less elegant, and also makes it no longer directly valid SLiM
code. But don’t worry, slimr knows how to transform this
syntax back into proper SLiM code, which you can see by printing out the
resulting slim_script object.
slim_script(
slim_block(initialize(),
{
## set the overall mutation rate
Init$initializeMutationRate(1e-7);
## m1 mutation type: neutral
Init$initializeMutationType("m1", 0.5, "f", 0.0);
## g1 genomic element type: uses m1 for all mutations
Init$initializeGenomicElementType("g1", m1, 1.0);
## uniform chromosome of length 100 kb
Init$initializeGenomicElement(g1, 0, 99999);
## uniform recombination along the chromosome
Init$initializeRecombinationRate(1e-8);
}),
slim_block(1,
{
sim%.%SS$addSubpop("p1", 500);
}),
slim_block(10000,
{
sim%.%SS$simulationFinished();
})
) -> script_2
script_2#> <slimr_script[3]>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 99999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }Okay, next up, let’s see some of the neat stuff we can do to
manipulate these SLiM scripts in R. First, we will see how to use R to
fill in parts of the SLiM script with data from R. This is accomplished
using the slimr_template function, which can be used to
insert R generated data into a SLiM script.
Templating
This is the simplest to get a SLiM script which has elements that can
be filled in later with a templating mechanism. So, how does that work?
Well, anywhere we want something in the SLiM script to be ‘in-fillable’,
that is, to contain a variable section of the script that we can
subsitute data into later, you can use the slimr_template
function in place of what was formerly a hard-coded value. Here is an
example using the same recipe as above, but in this case we want to be
able to fill-in the genome size and the muation rate later:
slim_script(
slim_block(initialize(),
{
## set the overall mutation rate (default = 1e-7)
initializeMutationRate( slimr_template("mut_rate", 1e-7) );
## m1 mutation type: neutral
initializeMutationType("m1", 0.5, "f", 0.0);
## g1 genomic element type: uses m1 for all mutations
initializeGenomicElementType("g1", m1, 1.0);
## uniform chromosome of length "genome_size" (default = 99999)
initializeGenomicElement(g1, 0, slimr_template("genome_size", 99999) );
## uniform recombination along the chromosome
initializeRecombinationRate(1e-8);
}),
slim_block(1,
{
sim.addSubpop("p1", 500);
}),
slim_block(10000,
{
sim.simulationFinished();
})
) -> script_temp
script_temp#> <slimr_script[3]>
#> block_init:initialize() {
#> initializeMutationRate(..mut_rate..);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, ..genome_size..);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#> This slimr_script has templating in block(s) block_init for
#> variables mut_rate and genome_size.The first argument to slimr_template provides a name for
the variable that will later be used to label values for replacement.
The second argument provides a default value. Anytime this script is
used without providing values for the templated variables, the default
values will be used. This argument is optional, and if not provided, an
error will be thrown if any variables have no values provided when later
using this script. The above printed script shows that it is templated
by printing a message describing the details below it, as well as
inserting the templated variables in the script with green
..x.., where x is the name of the variable.
Replacing values is now as simple as running
slim_script_render.
single_script <- slim_script_render(script_temp,
template = list(mut_rate = 1e-6, genome_size = 1999))
single_script#> <slimr_script[3]>
#> block_init:initialize() {
#> initializeMutationRate(1e-06);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 1999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }So we can see that we now have a slimr_script object
where our templated variables have been replaced!
slim_script_render can also render multiple different
values for the templated variables at the same time. To do this we can
just pass in a list of lists (or a data.frame), where each element at
the top level has a different set of values, and
slim_script_render will return a
slimr_script_coll object, which stands for a slimr script
collection. Let’s try it.
multi_script <- slim_script_render(script_temp,
template = list(
list(mut_rate = 1e-5, genome_size = 1999),
list(mut_rate = 1e-6, genome_size = 2999),
list(mut_rate = 1e-7, genome_size = 3999)
))
multi_script#> <slimr_script_coll[3]>
#> <1>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-05);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 1999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#>
#> <2>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-06);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 2999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#>
#> <3>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 3999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }To save typing you can also just include ..x..
specifications in the SLiM code and this will be recognized as a
templated variable. The disadvantage of this though is that you cannot
specify a default value, which can be pretty handy. Default values are
useful if, for example, you want to prototype a templated script, the
default values can serve as “test” values that will be filled in
automatically every time you want to test the script. Another potential
use is so that you only have to specify some of your variables in your
call to slim_script_render, in which case, anything you
don’t specify will take their default values. Additionally
slim_script_render has an argument that allows you to use
the default values in the case of a missing value in the data you supply
(such as in the data.frame style specification). Let’s look at an
example of both of these scenarios.
script_partial <- slim_script_render(script_temp,
template = list(
list(mut_rate = 1e-5),
list(genome_size = 2999)
))
script_partial#> <slimr_script_coll[2]>
#> <1>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-05);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 99999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#>
#> <2>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 2999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }Warnings are generated to help prevent unexpected behaviour. For
example if you simply misspelled one of the templated variable names in
the template, this would be replaced by the default value. And without
the above warnings you would never know that it happened. If you
intended to specify all templated variables but you get this warning,
check that the template you specified is correct! As we mentioned above,
using a data.frame as a template is also possible. Here is
an example.
script_temp_df <- slim_script_render(script_temp,
template = data.frame(
mut_rate = c(1e-5, NA, 1e-6, 1e-7),
genome_size = c(2000, 3000, NA, 5000)
)
)
script_temp_df#> <slimr_script_coll[4]>
#> <1>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-05);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 2000);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#>
#> <2>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 3000);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#>
#> <3>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-06);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 99999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#>
#>
#> <...>
#>
#> and 1 more.So in this case the default settings are to not replace NAs, because
in some cases NA may be a valid value you want to insert in a SLiM
script (rarely). You can change this behaviour by setting
replace_NAs = TRUE.
script_temp_df2 <- slim_script_render(script_temp,
template = data.frame(
mut_rate = c(1e-5, NA, 1e-6, 1e-7),
genome_size = c(2000, 3000, NA, 5000)
),
replace_NAs = TRUE)
script_temp_df2#> <slimr_script_coll[4]>
#> <1>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-05);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 2000);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#>
#> <2>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 3000);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#>
#> <3>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-06);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 99999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#>
#>
#> <...>
#>
#> and 1 more.Now all values have been replaced, either by their values in the
data.frame or by their defaults in the case that the
data.frame value was missing. This example also shows the
default print maximum for slimr_script_coll objects is 3.
To see the remaining slimr_script you can either use
print(script_temp_df2, max_show = 4) or you can subset in
the standard way, e.g:
script_temp_df2[3:4]#> <slimr_script_coll[2]>
#> <1>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-06);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 99999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }
#>
#> <2>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 5000);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:10000 early() {
#> sim.simulationFinished();
#> }If you want to check what the template settings for a
slimr_script are, use
slim_template_info().
slim_template_info(script_temp)
#> $block_init
#> $block_init$mut_rate
#> [1] 1e-07
#>
#> $block_init$genome_size
#> [1] 99999There is one additional argument to slimr_template that
we haven’t talked about yet, which is called
unquote_strings. This option allows you to insert unquoted
string values in a template. The default value, which is
FALSE, means that slimr_template assumes that
any variables of class “character” to be inserted are meant to be quoted
in the SLiM script, so they will be treated as literal strings by SLiM.
However, in some cases you may wish to use character values to insert
references to SLiM objects themselves, in which case, they must be
unquoted. As an example, perhaps you want to refer to different
Subpopulations in a SLiM script dynamically using templating. To do this
we can set unquote_strings = TRUE. As an example if we want
to refer to a Subpopulation dynamically, and we don’t use
unquote_strings = TRUE:
slim_script(
slim_block(initialize(),
{
## set the overall mutation rate (default = 1e-7)
initializeMutationRate(1e-7);
## m1 mutation type: neutral
initializeMutationType("m1", 0.5, "f", 0.0);
## g1 genomic element type: uses m1 for all mutations
initializeGenomicElementType("g1", m1, 1.0);
## uniform chromosome of length "genome_size" (default = 99999)
initializeGenomicElement(g1, 0, 99999);
## uniform recombination along the chromosome
initializeRecombinationRate(1e-8);
}),
slim_block(1,
{
sim.addSubpop("p1", 500);
sim.addSubpop("p2", 200);
}),
slim_block(1, .., late(), {
slimr_template("subpop", "p1")%.%.P$fitnessScaling = 0.5
}),
slim_block(10000,
{
sim.simulationFinished();
})
) -> script_q
script_q#> <slimr_script[4]>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 99999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> sim.addSubpop("p2", 200);
#> }
#>
#> block_3:1:10000 late() {
#> ..subpop....fitnessScaling = 0.5;
#> }
#>
#> block_4:10000 early() {
#> sim.simulationFinished();
#> }
#> This slimr_script has templating in block(s) block_3 for
#> variables subpop.If we try and replace the Subpopulation now, we will get invalid SLiM code:
slim_script_render(script_q, template = dplyr::tibble(subpop = c("p1", "p2")))
#> <slimr_script_coll[2]>
#> Error in parse(text = code, keep.source = TRUE): <text>:2:9: unexpected symbol
#> 1: {
#> 2: "p1"..fitnessScaling
#> ^Since this is also invalid R code, we get a parsing error. Now, if we
use unquote_strings = TRUE:
slim_script(
slim_block(initialize(),
{
## set the overall mutation rate (default = 1e-7)
initializeMutationRate(1e-7);
## m1 mutation type: neutral
initializeMutationType("m1", 0.5, "f", 0.0);
## g1 genomic element type: uses m1 for all mutations
initializeGenomicElementType("g1", m1, 1.0);
## uniform chromosome of length "genome_size" (default = 99999)
initializeGenomicElement(g1, 0, 99999);
## uniform recombination along the chromosome
initializeRecombinationRate(1e-8);
}),
slim_block(1,
{
sim.addSubpop("p1", 500);
sim.addSubpop("p2", 200);
}),
slim_block(1, .., late(), {
slimr_template("subpop", "p1", unquote_strings = TRUE)%.%.P$fitnessScaling = 0.5
}),
slim_block(10000,
{
sim.simulationFinished();
})
) -> script_uq
slim_script_render(script_uq, template = dplyr::tibble(subpop = c("p1", "p2")))#> <slimr_script_coll[2]>
#> <1>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 99999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> sim.addSubpop("p2", 200);
#> }
#>
#> block_3:1:10000 late() {
#> p1..fitnessScaling = 0.5;
#> }
#>
#> block_4:10000 early() {
#> sim.simulationFinished();
#> }
#>
#> <2>
#>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 99999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> sim.addSubpop("p2", 200);
#> }
#>
#> block_3:1:10000 late() {
#> p2..fitnessScaling = 0.5;
#> }
#>
#> block_4:10000 early() {
#> sim.simulationFinished();
#> }Now we get two slimr_scripts with correct SLiM
syntax!
Generating R-friendly Output
We can also use special slimr functions to make SLiM
generate output that can easily be read back into R as data. This works
best in conjunction with slimr, which can run SLiM and pull
any output into R while it is running. To do this, use the
slimr_output function inside the slimr code to
wrap any parts of it you want outputted. slimr_output is
context-aware; if you wrap a SLiM object, the object will be converted
into a string (using SLiM’s paste function) and then sent
to the output stream; if you wrap a SLiM output function (such as
outputFull or outputVCF), its output will be
sent directly to the output stream without conversion (since it is
already output strings). Let’s try adding some output to our SLiM
script.
slim_script(
slim_block(initialize(),
{
## set the overall mutation rate
initializeMutationRate(1e-7);
## m1 mutation type: neutral
initializeMutationType("m1", 0.5, "f", 0.0);
## g1 genomic element type: uses m1 for all mutations
initializeGenomicElementType("g1", m1, 1.0);
## uniform chromosome of length 100 kb
initializeGenomicElement(g1, 0, 99999);
## uniform recombination along the chromosome
initializeRecombinationRate(1e-8);
}),
slim_block(1,
{
sim.addSubpop("p1", 500);
}),
slim_block(1, .., late(),
{
## output sample of 10 individuals' genomes in VCF format
slimr_output(p1%.%.P$outputVCFSample(sampleSize = 10), name = "VCF");
catn("Another successful round of evolution completed!")
}),
slim_block(100,
{
sim.simulationFinished();
})
) -> script_w_out
script_w_out#> <slimr_script[4]>
#> block_init:initialize() {
#> initializeMutationRate(1e-07);
#> initializeMutationType("m1", 0.5, "f", 0);
#> initializeGenomicElementType("g1", m1, 1);
#> initializeGenomicElement(g1, 0, 99999);
#> initializeRecombinationRate(1e-08);
#> }
#>
#> block_2:1 early() {
#> sim.addSubpop("p1", 500);
#> }
#>
#> block_3:1:100 late() {
#> {p1..outputVCFSample(sampleSize = 10) -> VCF}
#> catn("Another successful round of evolution completed!");
#> }
#>
#> block_4:100 early() {
#> sim.simulationFinished();
#> }Now if you happen to have slimr installed and setup you
can run this script through slim and see what that output looks like. To
see examples of using slim_run, see the vignette titles
“Simple Example using Migration and Fst”.